This site is a project for Georgia Tech’s CS 4641.
Background
Scientists seek to identify protein targets associated with a disease and then develop a molecule that can modulate that protein target. To describe the biological activity of a given molecule or drug, scientists assign a label referred to as mechanism-of-action (MoA).
The current approach to identify Moa is to treat a sample of human cells with the drug and then analyze the cellular responses with algorithms that search for similar patterns in genomic databases, such as libraries of gene expression or cell viability patterns of drugs with known MoAs. However, this method is very costly and time-consuming.
Dataset Information
The dataset in this project combines gene expression and cell viability data. The data is based on a new technology that measures simultaneously human cells’ responses to drugs in a pool of 100 different cell types. In addition, it has MoA annotations for more than 5,000 drugs in this dataset which will act as the targets.
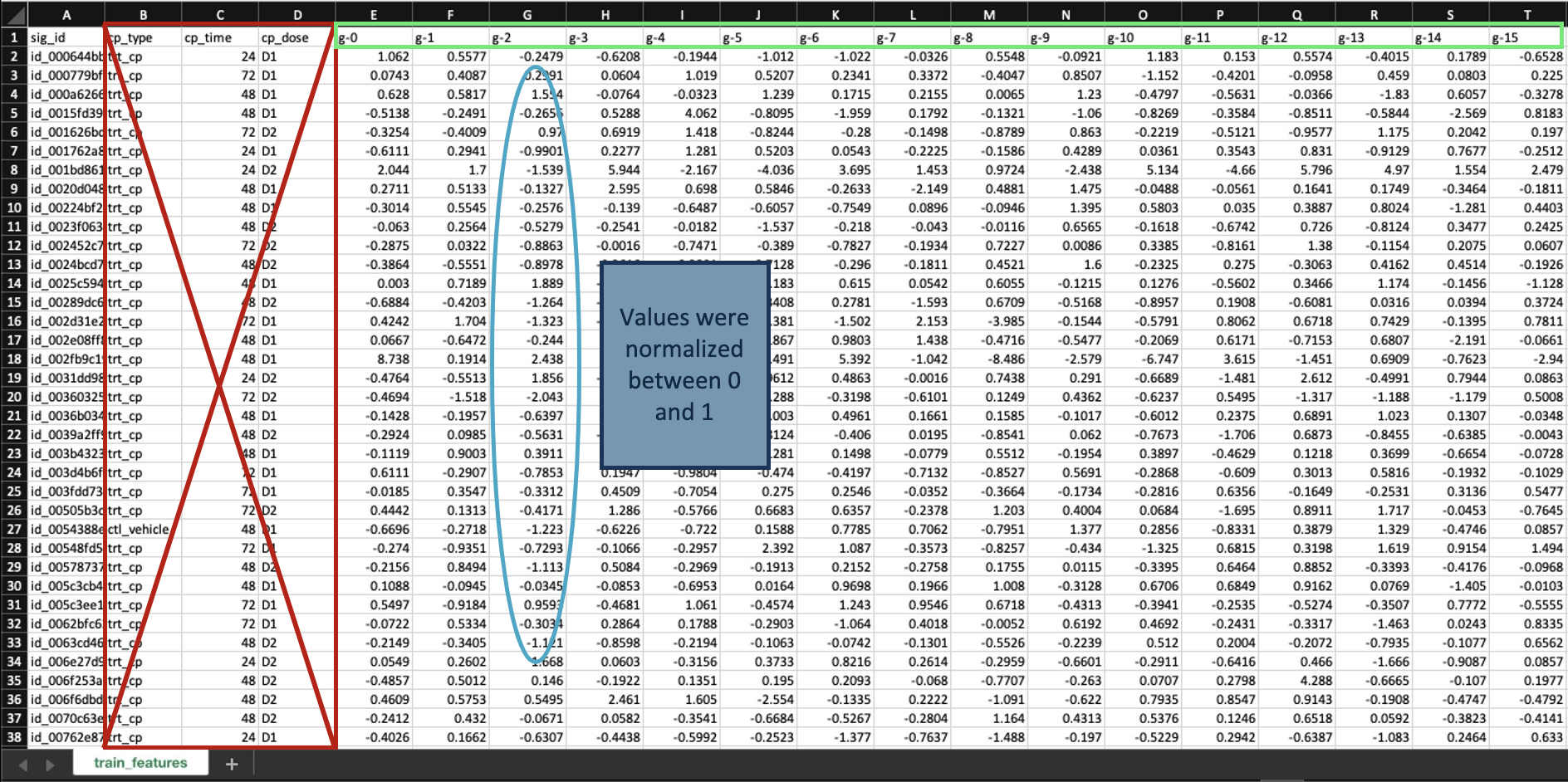
For our initial analysis, we discarded the dosage type and time because previous research studies indicate it should not significantly affect the MoAs present. Furthermore, the control vehicle is not needed because this data is already represented in the scored dataset on which our model will be trained. Finally, the remaining 852 columns representing gene expression values are normalized between 0 and 1 and used as a features.
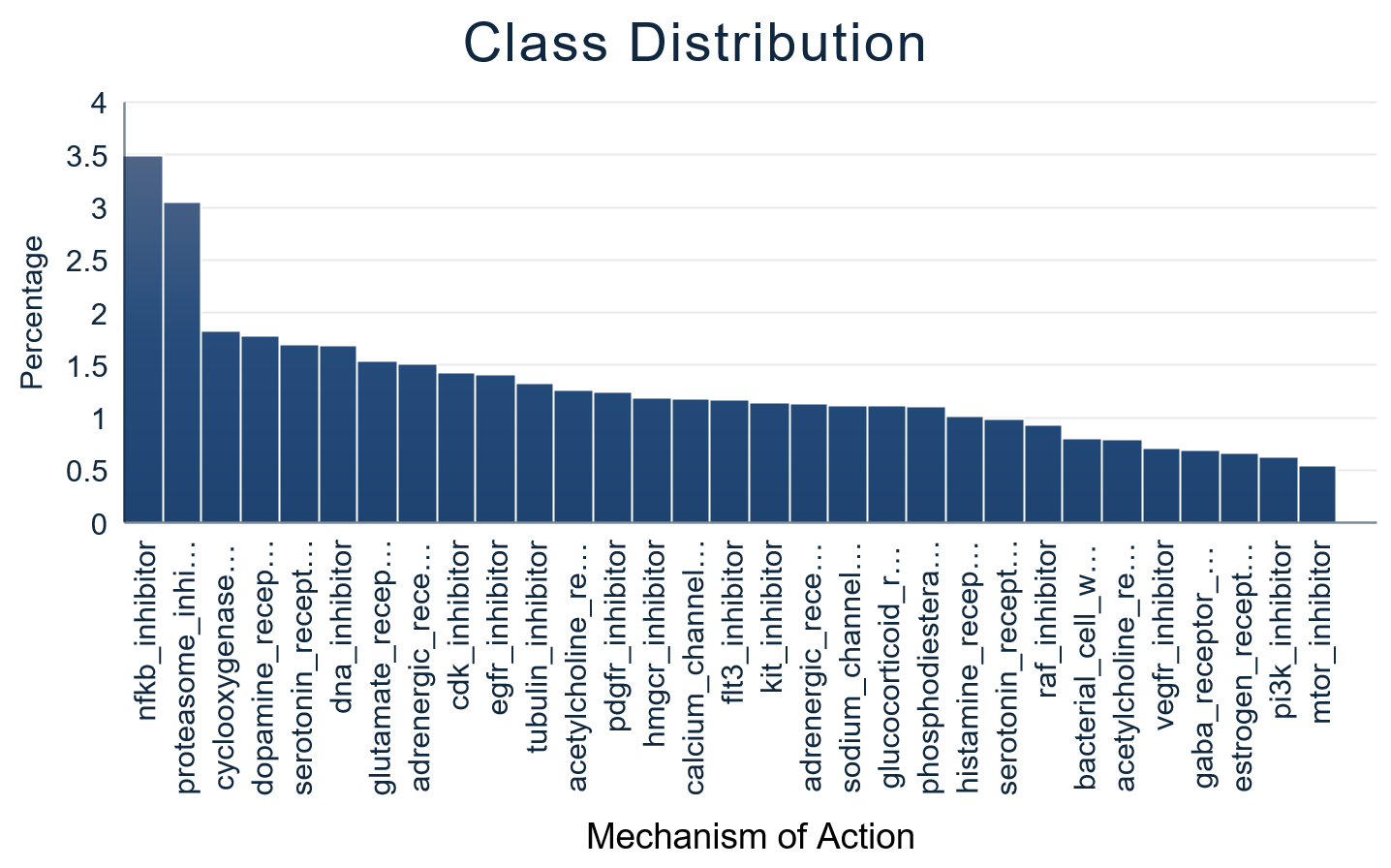
This figure shows the distributions of the top activated mechanisms of actions in our training data. The rest of the pathways only represent 30% of the total mechanisms. Note that not every mechanism of action is activated at any point, thus leading to a class distribution not summed to 100%. Based off the data set and our given time constraint, we didn’t manually identify any significant outliers. However, we plan on using our PCA algorithm with a parameter of 95% variance to not only reduce dimensionality but also remove the outliers from the dataset and thus ensure that they don’t affect our results negatively.
Methods
The Goal
Given various inputs such as gene expression and cell viability we hope to develop an algorithm that automatically labels each case in the test set as one or more MoA classes. Since drugs can have multiple MoA annotations, the task is a multi-label classification problem.
Unsupervised learning — PCA
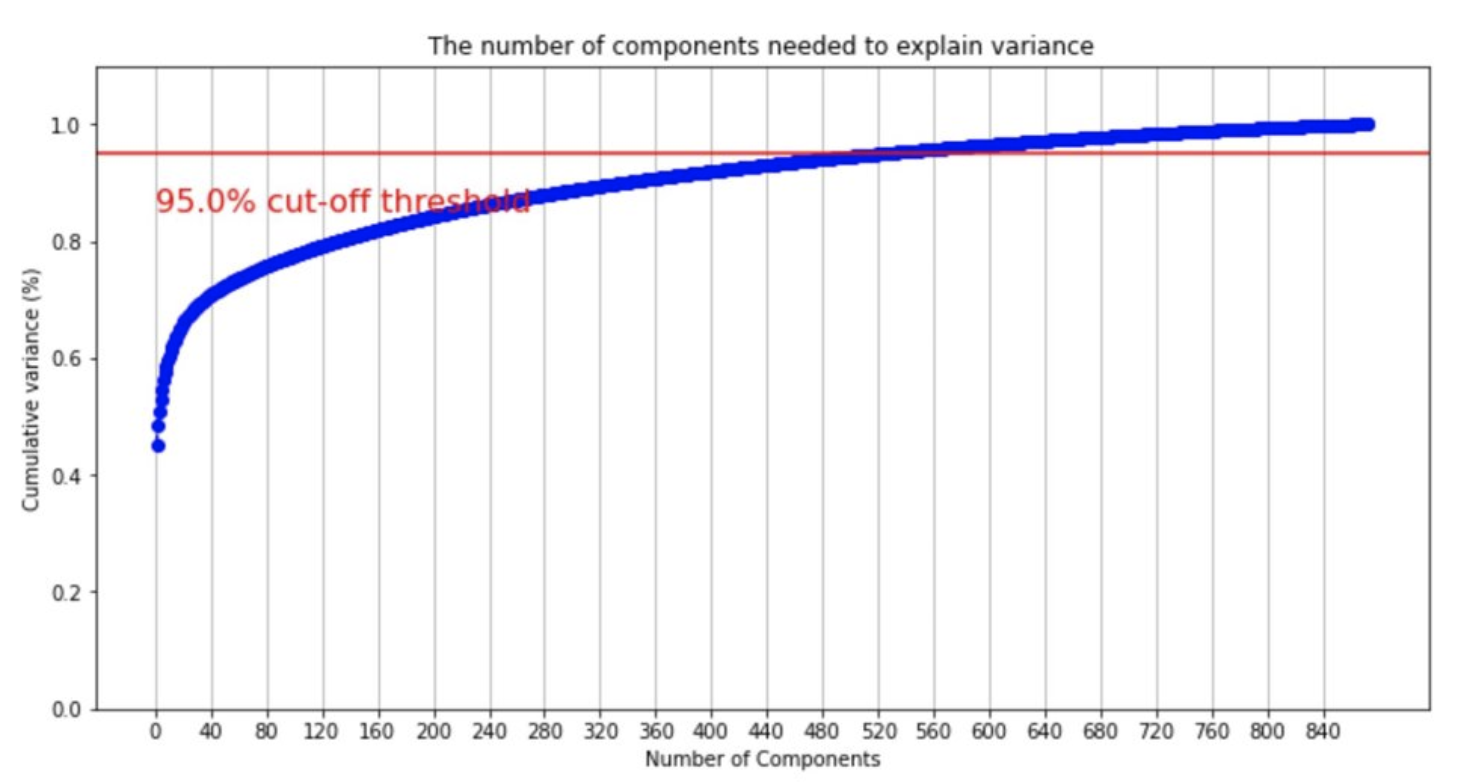
The 852 features in the dataset lead to a significant dimensionality, so, as stated before, we used PCA for dimensionality reduction. We determined that 95% is an optimal cut-off threshold and analyzed the number of components required to achieve that threshold. As shown in the figure above, the number of components needed to achieve 95% variance capture is about 525. After applying PCA, we have three different variations of the dataset to choose from: only using original data, only using PCA data, or using a combined dataset containing original and PCA data.
Supervised learning
Log Loss Equation
To determine the performance of our model in predicting the MoA of each sig_id, we will use the following log loss equation:
- N is the number of sig_id observations in the test data (i=1,…,N)
- M is the number of scored MoA targets (m=1,…,M)
- ŷ i, m is the predicted probability of a positive MoA response for a sig_id
- yi, m is the ground truth, 1 for a positive response, 0 otherwise
LG Boost
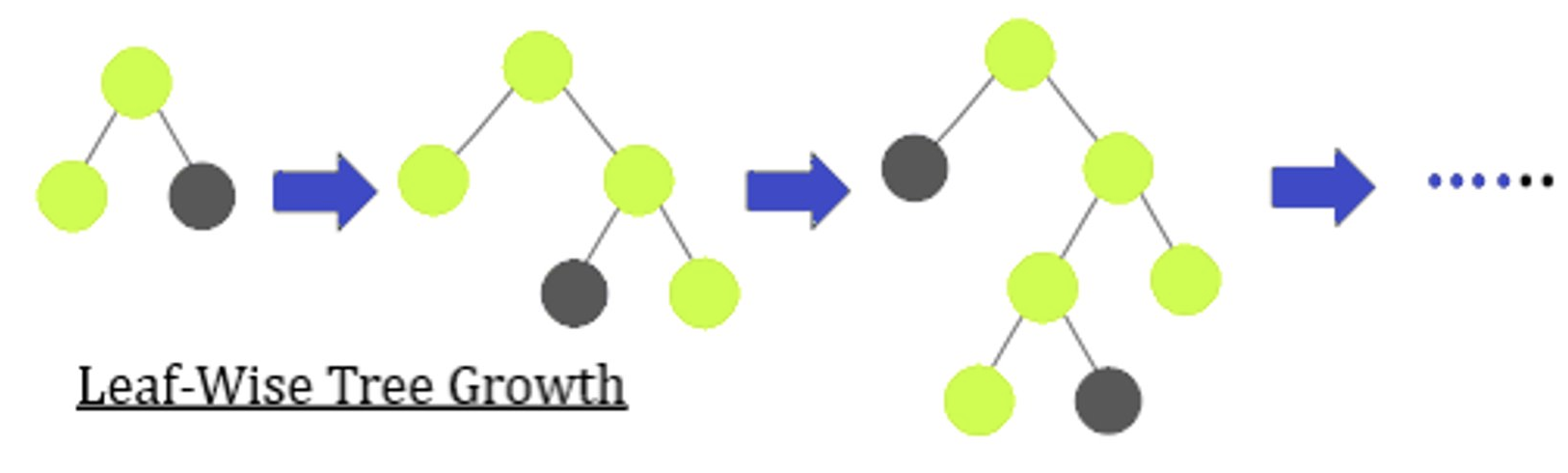
Initially, we wanted to use Extreme Gradient Boost because our problem space is multiclassification. However, because of the large dimensionality in the dataset, this was not something we could use. We also tried to tune the parameters and use a GPU, however, it still couldn’t handle this large dataset and would cause errors not having enough available RAM. We realized that we could use a Light Gradient Boost instead. In terms of parameters, with the help of previous work, we were able to tune the early stopping round parameter, learning rate, max depth and most importantly the number of leaves, which controls the complexity of the model. These are some of the more import parameters for this algorithm. We expected this to give suboptimal results, but we wanted to have a benchmark for later comparisons. This diagram shows how this algorithm works, splitting the tree leaf-wise. We also use a k-fold with five splits for most optimal results.
Neural Network
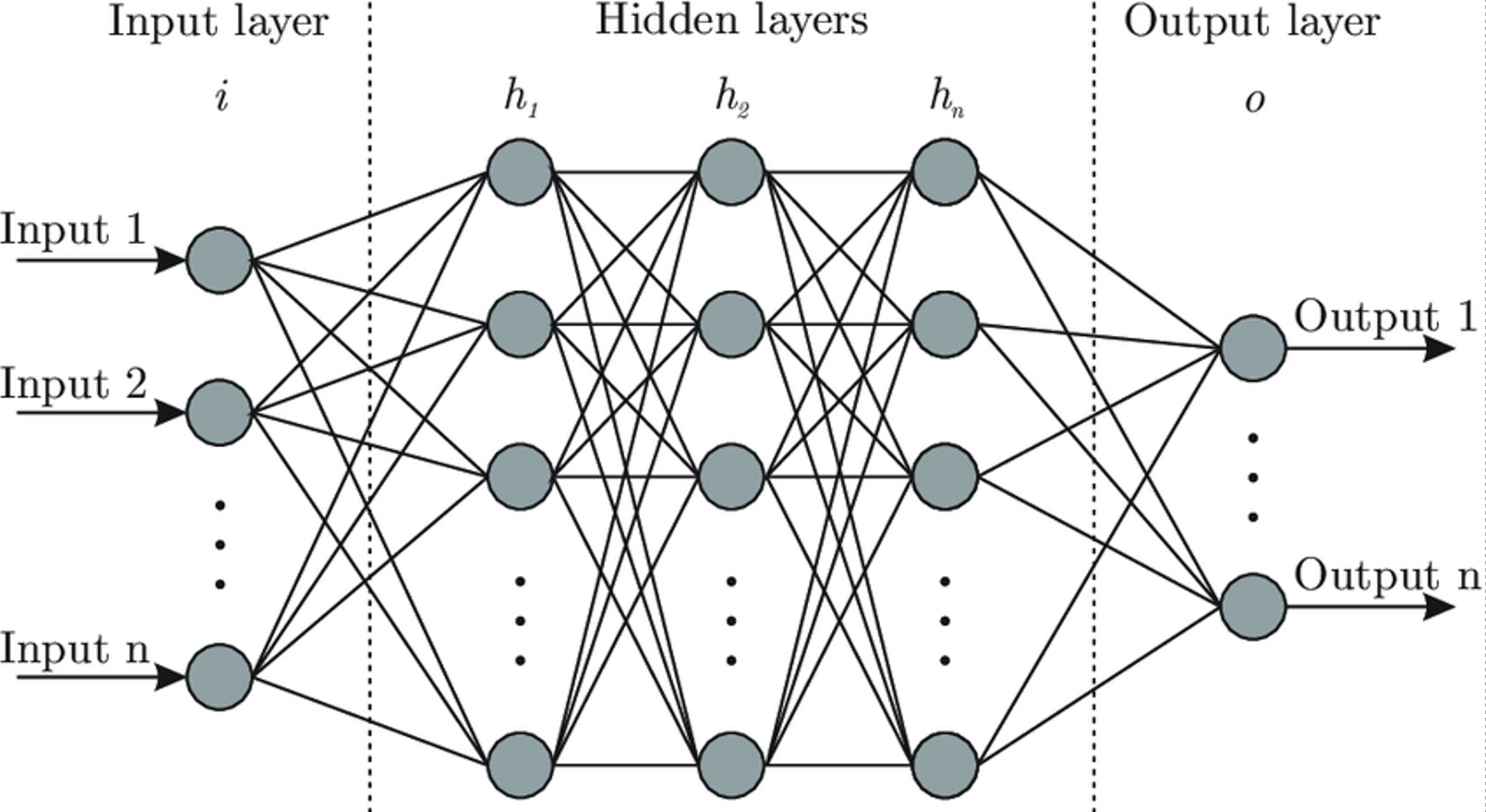
Next, we applies a neural network to the data. The neural network we used consisted of three dense layers with the first two layers having batch normalization and dropout after. This allows us to reduce overfitting. The activation function for both layers was the rectified linear unit (ReLU). The final layer has a sigmoid activation. The optimization function we used was ADAM. A grid search was used to find optimal parameters for the model. Parameters include learning rate, number of nodes in each layer, etc. A KFold with five splits was used to make sure model performances were accurate.
Results
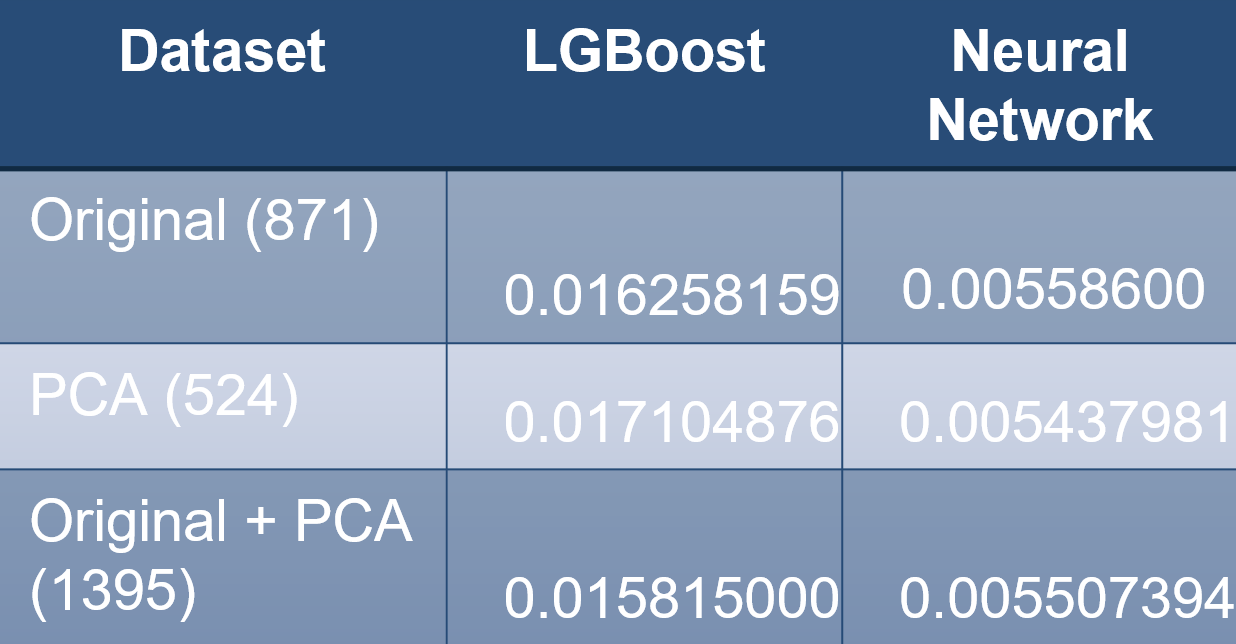
Based on the MoA annotations, the accuracy of solutions will be evaluated on the average value of the logarithmic loss function applied to each drug-MoA annotation pair. We expected the neural network 5o outperform the LGBoost model, which it did. Moreover, is seems like the neural network with only PCA data did marginally better than the other datasets. For perspective on the results, our scores are in the top 10% of Kaggle submissions.

For reference, this is a sample output that our model produced. this table shows the probabilities of each drug having a certain mechanism of action. The rows are each drug and the columns are each mechanism of action. So for example, probabilities in red, like 1 and 0.99, indicate that the drug’s mechanism of action is whichever one is in that column.
Following our experiments and comparing log loss values, we conclude that using PCA data with the neural network provides the best overall prediction performance. However, we are limited by the processing power available and limited access to GPU. In addition, we removed categorical variables from our dataset to decrease the difficulty of the learning model. Overall, We believe that our work could contribute to predicting a compound’s MoA given its cellular signature in broader scenarios, thus helping scientists advance the drug discovery process.formance. Our work could contribute to classification of current and future drugs to help doctors discover effective treatments.
References
-
Peach KC, Bray WM, Winslow D, Linington PF, Linington RG. Mechanism of action-based classification of antibiotics using high-content bacterial image analysis. Mol Biosyst. 2013 Jul;9(7):1837-48. doi: 10.1039/c3mb70027e. Epub 2013 Apr 23. PMID: 23609915; PMCID: PMC3674180.
-
Eltze M. Multiple mechanisms of action: the pharmacological profile of budipine. J Neural Transm Suppl. 1999;56:83-105. doi: 10.1007/978-3-7091-6360-3_4. PMID: 10370904.
-
“Mechanisms of Action (MoA) Prediction.” Kaggle, www.kaggle.com/c/lish-moa/overview.